hemophilia - Coagulation cascade
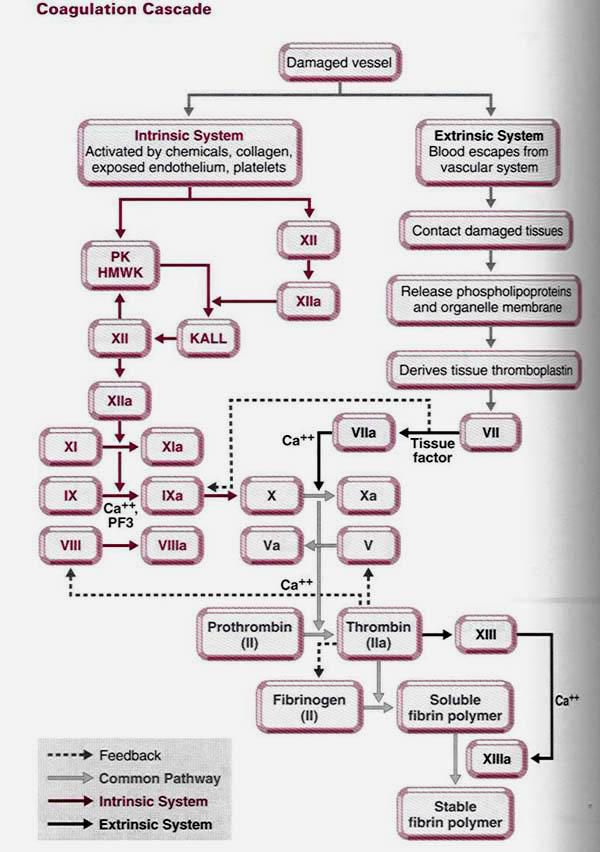 . .
Initial pathway (Extrinsic)
1. A cell membrane protein called tissue factor (TF), present on the outside of all human cells with the exception of red blood cells and endothelium, binds with a plasma protein, Factor VII (FVII) converting FVII to the active FVIIa.
This initial pathway is independent of Factor VIII (factor missing in hemophilia A) and Factor IX (factor missing in hemophilia B). The question arises, why can’t the body utilize only this pathway in the absence of Factor VIII or IX?
When the body has made a small amount of fibrin, a substance known as Tissue Factor Pathway Inhibitor (TFPI) is released. This inhibitor binds to the TF:FVIIa/FXa complex, preventing further formation of factor FXa. It is thought that TFPI is released to protect against overreation of the coagulation system. At this point, the intrinsic pathway is activated.
II. Intrinsic Pathway
|